Portal del Medicamento
Esta tabla incluye el cuerpo de datos relativo al portal, es decir información relativa a contenidos, mapa web, etc. en función de la página en la que se encuentre el usuario.

Medicamentos biosimilares, despejando dudas
Ojo de Markov número treinta y seis - abril 2015
Según el IMS Health 8 de los 10 medicamentos con mayor gasto en Europa en 2013 fueron biológicos. La patente de algunos de estos medicamentos como rituximab y trastuzumab, entre otros, ya ha expirado y en este año y hasta 2020 ocho medicamentos biológicos más perderán la exclusividad de mercado. Los análisis realizados por el IMS Health han estimado el mercado global de estos fármacos en 72.000 millones de $ americanos (67.000 millones de euros) y el de España en 1.500 millones de euros. Ante este escenario, existe un enorme interés en obtener biosimilares y acceder así a este mercado tan goloso que, previsiblemente, tendrá un elevado impacto económico en los próximos años.
8 de los 10 medicamentos con mayor gasto en Europa en 2013 fueron biológicos. La patente de algunos de estos medicamentos como rituximab y trastuzumab, entre otros, ya ha expirado y en este año y hasta 2020 ocho medicamentos biológicos más perderán la exclusividad de mercado. Los análisis realizados por el IMS Health han estimado el mercado global de estos fármacos en 72.000 millones de $ americanos (67.000 millones de euros) y el de España en 1.500 millones de euros. Ante este escenario, existe un enorme interés en obtener biosimilares y acceder así a este mercado tan goloso que, previsiblemente, tendrá un elevado impacto económico en los próximos años.
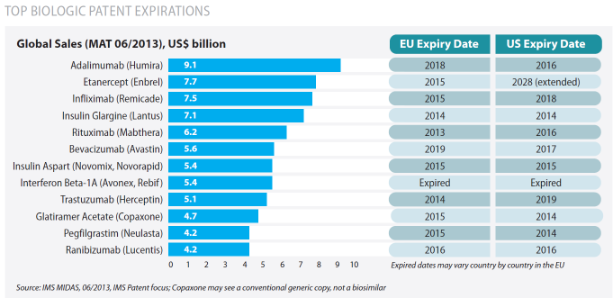
Aunque en nuestro país se utilizan biosimilares desde 2007, año en que se comercializó la somatropina, la salida al mercado del primer anticuerpo monoclonal biosimilar, infliximab, y la proyección de que la patente de varios medicamentos biológicos está a punto de expirar ha provocado una gran polémica. En el momento actual, al igual que ocurrió en su día con los genéricos, el debate está en auge y las jornadas y reuniones sobre biosimilares, con enfoques muy distintos, se suceden continuamente. Los argumentos científicos, las posiciones y normas reguladoras están teniendo diferentes interpretaciones y, por tanto, se está originando cierta confusión. Con el fin de esclarecer los aspectos legales y científicos más relevantes sobre estos medicamentos, Sacyl ha elaborado esta entrada para los profesionales que recoge los puntos más debatidos sobre los fármacos biosimilares.
Un biosimilar es un medicamento biológico similar a otro ya existente (el medicamento de referencia o innovador) que se desarrolla de acuerdo a estrictos requisitos exigidos por la Agencia Europea del Medicamento (EMA).
Se entiende por medicamento biológico el que contiene uno o más principios activos sintetizados o derivados de una fuente biológica (fluidos o tejidos humanos o animales; microorganismos…) Algunos ejemplos son la insulina, las eritropoyetinas o los anticuerpos monoclonales. Estos medicamentos pueden ser producidos por diferentes técnicas:
- Directamente por el organismo vivo, como los factores de coagulación extraídos y purificados de plasma humano.
- Por biosíntesis en células vegetales o animales, bacterias, levaduras y virus mediante técnicas de biología molecular o biotecnológicas que implican la manipulación del material genético. A los medicamentos obtenidos de este modo se les denomina medicamentos biotecnológicos.
Un biosimilar es un medicamento biológico más
Desarrollo de un fármaco biotecnológico
La mayoría de medicamentos biotecnológicos son proteínas grandes y complejas cuyo desarrollo es complicado. Su proceso de fabricación, sean biosimilares o no, es muy sensible y pequeños cambios pueden dar lugar a diferencias mínimas en su estructura, de ahí que se suela decir que "el proceso es el producto".
Estas variaciones estructurales entre dos fármacos biotecnológicos de una misma molécula se pueden producir entre dos innovadores obtenidos en distintas líneas celulares, entre un fármaco antes y después de hacer modificaciones en su proceso de fabricación, y también entre un biosimilar y su innovador.
¿Importa la variabilidad estructural si se demuestra la similitud terapéutica?
Por supuesto que no. Lo que es importante es evaluar en qué medida las posibles diferencias estructurales de los medicamentos biológicos afectan a la calidad, eficacia y seguridad del fármaco. De hecho, la EMA no exige realizar siempre ensayos clínicos cuando se producen modificaciones estructurales en un producto debido a cambios en el proceso de fabricación sino que esto se decidirá para cada caso en particular (ver guía sobre productos biológicos/biotecnológicos sujetos a cambios en su proceso de fabricación).
La variabilidad entre biosimilar e innovador es irrelevante si existe similitud terapéutica
Comparabilidad y biosimilitud
En el documento informativo de consenso "What you need to know about biosimilar Medicinal Products" publicado por la Comisión Europea se explican muy bien estos conceptos. Comparabilidad es un concepto científico que se refiere al ejercicio de comparar todos los aspectos moleculares y clínicos entre dos productos biológicos. Biosimilitud es el término legal utilizado en la Unión Europea que expresa la comparabilidad entre un biosimilar y el producto de referencia.
La "Guideline on similar biological medicinal products" de la EMA establece de modo general los principios necesarios para demostrar la comparabilidad entre dos medicamentos biológicos (bien sea biosimilar e innovador o dos biológicos entre sí). Se recomienda lo siguiente:
1º. Mediante datos preclínicos demostrar que la calidad de ambos productos es similar. Aportar datos comparativos de la caracterización molecular y biológica (estructura tridimensional, actividad biológica, etc.) de los dos productos.
2º. Si los datos in vitro indican que no existen diferencias relevantes entre ambos productos se procederá a la evaluación clínica. Se realizarán ensayos clínicos comparativos suficientemente sensibles en diseño, población de estudio y variables para detectar las posibles diferencias. Estos estudios servirán para garantizar que la eficacia y seguridad del biosimilar es adecuada.
Señalar que los criterios de comparabilidad que la EMA exige a los biosimilares son superiores a los solicitados a los medicamentos de referencia. El biosimilar debe ser comparado directamente con el innovador a lo largo de todo su desarrollo y se le exige estudios de no inferioridad frente al de referencia mientras que al innovador solo se le exige estudios frente a placebo. Expertos en el tema han afirmado que "algunos medicamentos biológicos originales como ciertos interferones no aguantarían los exámenes de comparabilidad que se exige a los biosimilares".
La EMA exige a los biosimilares ensayos clínicos comparativos frente al producto de referencia para demostrar que ambos productos son similares en eficacia y seguridad
Inmunogenicidad
La capacidad para inducir respuesta inmune o inmunogenicidad es inherente a cualquier medicamento biológico. Depende de numerosos factores como la genética del individuo, el tipo de enfermedad, el proceso de producción, el tipo de molécula, etc. Las limitaciones del proceso de producción de los medicamentos biotecnológicos que impiden obtener un producto idéntico al original han sido utilizadas para poner en duda la comparabilidad de los medicamentos biosimilares en cuanto a su potencial inmunógeno. En esta línea, la EMA dispone de directrices específicas de las proteínas terapéuticas (adoptadas en 2006 y revisada en 2013) y también de los anticuerpos monoclonales que describen como se debe evaluar la inmunogenicidad de estos productos, bien sean innovadores o biosimilares. En el caso de los biosimilares, la normativa contempla que la inmunogenicidad del biosimilar y del producto de referencia debe ser comparada tanto in vitro como en ensayos clínicos.
La inmunogenicidad de los medicamento biotecnológicos es un aspecto que preocupa seriamente a la EMA porque puede afectar tanto a la seguridad como a la pérdida de eficacia (neutralización del fármaco por anticuerpos). Prueba de ello es la interrupción de ensayos clínicos por detectar inmunogenicidad como ocurrió en 2009 con un ensayo de eritropoyetina biosimilar o el hecho de que en el plan de gestión de riesgos post-autorización de todos los medicamentos biológicos se incluya vigilar y revisar los datos registrados sobre reacciones inmunológicas.
Conforme a las normas comunitarias y nacionales todos los medicamentos nuevos, incluidos los biosimilares o cualquier otro biológico, son identificados con un símbolo (triángulo negro) durante los 5 primeros años, con el objeto de hacer un especial seguimiento y comunicación de los efectos adversos.
La inmunogenicidad es evaluada tanto in vitro como en ensayos clínicos y es una cuestión de extremada importancia en todos los medicamentos biológicos, por ello la EMA les exige un plan de gestión de riesgos
Extrapolación de indicaciones
La experiencia clínica acumulada con los medicamentos biotecnológicos originales debe simplificar la investigación y desarrollo del biosimilar. Por ello, en ciertos casos, tanto la EMA como la FDA permiten extrapolar al biosimilar, algunas o todas las indicaciones del medicamento de referencia sin la necesidad de llevar a cabo ensayos clínicos. La extrapolación de indicaciones requiere una adecuada justificación científica y ejercicio de comparabilidad (por ejemplo: demostrar que el mecanismo de acción y / o el receptor que participan en las indicaciones extrapoladas son el mismo). Este aspecto se contempla en las guías específicas para cada biosimilar o en el Informe Público de Evaluación Europeo (EPAR) del biosimilar en cuestión. Un ejemplo es el EPAR de infliximab biosimilar que dice textualmente: "la extrapolación de los datos farmacocinéticos, de eficacia y seguridad generados en los dos ensayos clínicos, en espondilitis anquilosante y en artritis reumatoide, a otras indicaciones de Remicade®, incluyendo enfermedad inflamatoria intestinal, se considera posible en base a los resultados de la exhaustiva comparabilidad in vitro e in vivo". Además, el CHMP considera que las nuevas evidencias genéticas e histológicas que establecen vinculaciones clínicas e histológicas entre la inflamación intestinal, la espondilitis anquilosante y la enfermedad de Crohn apoyan esta decisión. El EPAR de infliximab también recoge que "los datos clínicos preliminares de una pequeña cohorte de pacientes de Corea del Sur con enfermedad de Crohn y colitis ulcerosa indican una respuesta similar del biosimilar de infliximab en comparación con los datos registrados de Remicade®".
La EMA evalúa cada fármaco minuciosamente y si existe una adecuada justificación científica puede otorgar a los medicamentos biosimilares las mismas indicaciones que tiene el medicamento de referencia
Intercambiabilidad. Marco legal en España
La EMA no hace referencia a la intercambiabilidad y sustitución de medicamentos biológicos, y deja este aspecto a criterio de las autoridades sanitarias de cada país miembro de la UE.
En España las restricciones a la sustitución de unos medicamentos por otros (orden SCO/2874/2007) se ciñen a un grupo de productos entre los cuales están los fármacos biológicos. Esto aplica a la sustitución por el farmacéutico, recogida en el capítulo IV Del uso racional de medicamentos en las oficinas de farmacia de la Ley 29/2006
En el ámbito hospitalario es posible un mayor seguimiento y monitorización de los tratamientos, por lo que el uso de biosimilares en lugar de biológicos originales se puede realizar con mejor garantía de seguridad. Cada centro decidirá acerca de los criterios de utilización de los biosimilares, al igual que del resto de fármacos de ámbito hospitalario, cuando no exista una comisión específica para la selección de medicamentos a nivel nacional o autonómico.
En el ámbito hospitalario el uso de biosimilares en lugar de biológicos originales debe realizarse de forma consensuada y con garantías de seguridad y seguimiento individualizado
El mercado de los biosimilares el hoy y el mañana…
En Europa, hasta ahora la utilización de biosimilares no ha cumplido con las expectativas deseadas. El acceso de los biosimilares al mercado se ha visto frenado por diversas causas:
- Las elevadas exigencias científicas y técnicas para su desarrollo, como la necesidad de ensayos clínicos para demostrar la biosimilitud o el complejo proceso de fabricación.
- La lentitud de la EMA para otorgar la autorización y la legislación en cuanto a la intercambiabilidad de los diferentes estados miembros.
- La falta de experiencia de los profesionales sanitarios y el temor injustificado de que los biosimilares sean inferiores en eficacia o seguridad a los originales.
- La presión de la industria propietaria de los innovadores para desacreditar a los biosimilares.
En la actualidad en Europa hay 19 medicamentos biosimilares autorizados: 8 filgastrim, 5 epoetinas, 2 hormonas folículo estimulantes, 2 infliximab, 1 hormona de crecimiento y 1 insulina glargina. Como ya se ha comentado, en los próximos años expirará la patente de muchos medicamentos biológicos y es previsible que el mercado de los biosimilares crezca significativamente.
En España los medicamentos biosimilares están incluidos dentro del sistema de precios de referencia (art 93 de Ley 29/2006). El Ministerio de Sanidad ha fijado las reducciones de precio para los medicamentos biosimilares en un 30% (esta información no se ha concretado normativamente), por lo que su utilización supondrá un enorme ahorro para nuestro sistema de salud que incidirá fundamentalmente en el gasto hospitalario y de forma global en la sostenibilidad del Sistema Nacional de Salud (SNS).
La utilización de fármacos biosimilares ayudará a la sostenibilidad del SNS, de forma que facilitará la incorporación de otros fármacos innovadores

Autora: Belén Calabozo FreileRevisores: Tomás Caro Patón; Judit Ceruelo Bermejo, Sagrario Garrido López, Laura Isusi Lomas,
Natalia Revilla CuestaÚltima modificación: septiembre 2016